Митохондриальные заболевания
Митохондриальные заболевания (МЗ) — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках.
Историческая справка:
Понятие «митохондриальные болезни» сформировалось в медицине в конце ХХ века. В первую очередь были изучены болезни, связанные с мутациями митохондриальной ДНК, открытой в начале 60-ых годов. Полная первичная структура митохондриальной ДНК человека была опубликована в 1981 го¬ду и уже в конце 80-ых годов была доказана ведущая роль ее мутаций в развитии ряда наследственных заболеваний. К последним относятся: наследственная атрофия зрительных нервов Лебера, синдром NARP (нейропатия, атаксия, пигментный ретинит), синдром MERRF (миоклонусэпилепсия с «рваными» красными волокнами в скелетных мышцах), синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды), синдром Кернса-Сейра (пигментный ретинит, наружная офтальмоплегия, блокада сердца, птоз, мозжечковый синдром), синдром Пирсона (поражение костного мозга, панкреатическая и печеночная дисфункции) и многие другие.
В меньшей степени изучены наследственные митохондриальные дефекты, связанные с повреждением ядерного генома.
Патогенез.
Митохондрии отвечают за выработку большей части энергии, необходимой для функционирования клеток. Фактически они являются настолько важным источником энергии, что в каждой клетке их сотни. При МЗ могут «выключиться» как часть митохондрий, так и все они, что приводит к прекращению выработки необходимой энергии
Поскольку наиболее энергоемкими являются нервные и мышечные клетки, при МЗ наиболее распространены мышечные и неврологические проблемы, такие, как мышечная слабость, непереносимость физических нагрузок, потеря слуха, нарушения баланса и координации, эпиприступы.
Когда клетка заполнена дефектными митохондриями, она не только лишена АТФ, но в ней могут накапливаться неиспользуемые молекулы топлива и кислород, что приводит к катастрофическим последствиям. В этом случае избыточные молекулы топлива используются для синтеза АТФ неэффективно, в результате чего могут образовываться потенциально опасные продукты, такие, как молочная кислота (Это также происходит, когда клетки испытывают недостаток кислорода, например – мышечные клетки при усиленных физических нагрузках). Накопление молочной кислоты в крови – лактатацидоз – ассоциировано с мышечной усталостью, и может вызывать повреждение нервной и мышечной тканей.
При этом неиспользуемый в клетке кислород может трансформироваться в разрушительные соединения, именуемые реактивными формами кислорода, включая т. н. свободные радикалы (Они являются мишенью для т. н. антиоксидантных препаратов и витаминов).
Синтезированная в митохондриях АТФ – основной источник энергии для сокращения мышечных и возбуждения нервных клеток (т. к. клетки этих тканей наиболее метаболически активны, энергетически зависимы). Таким образом, нервные и мышечные клетки особенно чувствительны к дефектам митохондрий. Комбинированный эффект от потери энергии и накопления токсинов в этих клетках, надо полагать, и вызывает развитие симптомов митохондриальных миопатий и энцефаломиопатий
Клиника
Характерные признаки митохондриальных цитопатий:
•скелетные мышцы: низкая толерантность к физической нагрузке, гипотония, проксимальная миопатия, включающая фациальные и фарингеальные мышцы, офтальмопарез, птоз
•сердце: нарушения сердечного ритма, гипертрофическая миокардиопатия
•центральная нервная система: атрофия зрительного нерва, пигментная ретинопатия, миоклонус, деменция, инсультоподобные эпизоды, расстройства психики
•периферическая нервная система: аксональная нейропатия, нарушения двигательной функции гастроинтестинального тракта
•эндокринная система: диабет, гипопаратиреоидизм, нарушение экзокринной функции панкреас, низкий рост
Таким образом, типичны для митохондриальных заболеваний вовлеченность разных органов и одновременное проявление внешне не связанных между собой аномалий. Примерами служат:
1. Мигрени с мышечной слабостью
2. Наружная офтальмоплегия с нарушением проводимости сердечной мышцы и мозжечковой атаксией
3. Тошнота, рвота с оптической атрофией и кардиомиопатией
4. Низкорослость с миопатией и инсультоподобным и эпизодами
5. Экзокринная дисфункция поджелудочной железы с сидеробластной анемией
6. Энцефало- миопатия с диабетом
7. Диабет с глухотой
8. Глухота с наружной офтальмоплегией, птозом и ретинопатией
9. Задержка развития или потеря навыков и офтальмоплегия, офтальмопарез
Характер и тяжесть клинических проявлений митохондриальных болезней определяется:
• тяжестью мутации мтДНК;
• процентным содержанием мутантной мтДНК в конкретных органах и тканях;
• энергетической потребностью и функциональным резервом органов и тканей, содержащих мтДНК (их “порогом чувствительности” к дефектам окислительного фосфори лирования).
Миопатия
Основные симптомы митохондриальной миопатии – истощение мышц и их слабость, и непереносимость физических нагрузок.
У некоторых индивидов слабость наиболее выражена в мышцах, контролирующих движения глаз и век. Два наиболее частых следствия такой слабости – это постепенный паралич движения глаз (прогрессирующая наружная офтальмоплегия, ПНО), и опущение верхних век (птоз). Зачастую люди автоматически компенсируют ПНО движениями головы для того, чтобы смотреть в различных направлениях, и могут даже не подозревать о каких либо проблемах. Птоз потенциально более неприятен, поскольку может ухудшить зрение, а также придает лицу апатичное выражение, но он может быть скорректирован хирургическим путем, либо использованием специальных очков с устройством для подъема века
Митохондриальные миопатии могут также вызывать слабость других мышц лица и шеи, что приводит к заплетающейся речи и трудностям с глотанием. В этих случаях могут помочь речевая терапия (занятия с логопедом) или включение в рацион питания таких продуктов, которые легче проглатываются.
Энцефаломиопатия
Митохондриальная энцефаломиопатия, как правило, включает некоторые из вышеупомянутых симптомов миопатии, дополненными одним или несколькими неврологическими симптомами. Также как и при миопатии, наблюдается значительная вариабельность симптомов обоего типа и тяжести течения у разных индивидов.
Среди наиболее частых симптомов митохондриальной энцефаломиопатии – нарушения слуха, мигренеподобные головные боли и эпиприступы. По крайней мере, в одном синдроме головные боли и эпиприступы часто сопровождается инсультоподобными эпизодами
Дополнительно к поражению глазных мышц, митохондриальная энцефаломиопатия может поражать как сами глаза, так и участки головного мозга, ответственные за зрение. Например, потеря зрения вследствие оптической атрофии (дегенерации зрительного нерва) или ретинопатии (дегенерации некоторых клеток, выстилающих глазное дно) – обычные симптомы митохондриальной энцефаломиопатии. По сравнению с мышечными проблемами, эти эффекты с большей вероятностью приводят к серьезным нарушениям зрения
Довольно часто митохондриальная энцефаломиопатия вызывает атаксию, или сложности с балансом и координацией.
Диагностика.
Ни один из отличительных симптомов митохондриального заболевания – мышечная слабость, непереносимость нагрузок, ухудшение слуха, атаксия, эпиприступы, неспособность к обучению, катаракта, диабет и низкорослость – не является уникальным именно для такого заболевания. Однако комбинация трех или более из этих симптомов у одного индивида свидетельствует в пользу митохондриального заболевания, особенно если симптомы затрагивают более одной системы организма
Физикальное обследование обычно включает в себя тесты на силу и выносливость, такие например, как повторяющиеся сжатия-разжатия кулака, или подъем и спуск по небольшой лестнице. Неврологическое обследование может включать в себя проверку рефлексов, зрения, речи и базовых когнитивных способностей.
Существует ряд рутинных клинических методов исследования, которые можно использовать при подозрении на митохондриальную цитопатию:
•лактатный ацидоз является практически постоянным спутником митохондриальных болезней (только этот признак является недостаточным для постановки диагноза, так как он может выявляться и при других патологических состояниях; в этом отношении может быть полезным измерение уровня лактата в венозной крови после умеренной физической нагрузки, например на велоэргометре)
•ЭЭГ – данные ЭЭГ не является достаточно специфическими
Образцы мышечных биоптатов целесообразно делить на три части – одна для микроскопического исследования (гистология, гистохимия и электронная микроскопия), вторая для энзимологического и иммунологического анализа (изучение характеристик компонентов дыхательной цепи) и третья – непосредственно для молекулярно-генетического анализа. Поиск известных мутаций на мышечном материале позволяет в большинстве случаев успешно осуществлять ДНК-диагностику болезни. При отсутствии из вестных мутаций мтДНК в мышечной ткани следующим этапом является развернутый молекулярно-генетический анализ – секвенирование всей цепи мтДНК (или кандидатных генов ядерной ДНК) с целью выявления нового варианта мутации.
Лечение.
Что касается терапии митохондриальных цитопатий, то речь может идти пока только о симптоматической.
Лечение митохондриальных болезней проводится обычно по двум основным направлениям:
•предупреждение повреждения митохондриальных мембран свободными радикалами с помощью антиоксидантов (витамин Е, a-липоевая кислота) и мембранопротекторов.
В практику входят всё новые препараты комбинированного действия, такие, например, как идебенон (Нобен) – улучшенный структурный аналог коэнзима Q10, благоприятно влияющий на активность дыхательного пути и обладающий выраженным антиоксидантным, антиапоптотическим и нейротрофическим действием.
Очевидно, что расширение терапевтического арсенала при митохондриальных болезнях диктует настоятельную необходимость того, чтобы практические врачи различных специальностей (неврологи, психиатры, педиатры, генетики, гематологи и др. ) были хорошо знакомы с алгоритмом диагностики этих заболеваний.
Как усилить работу митохондрий
Митохондрии представляют собой внутриклеточные органеллы эукариот, основной функцией которых является выработка АТФ в результате реакции окислительного фосфорилирования. (Logan, 2006)
Каждая митохондрия содержит высокоспециализированные мембраны, играющие ключевую роль в ее активности. Мембраны образуют два изолированных митохондриальных компартмента: внутренний матрикс и узкое межмембранное пространство. Каждый отдел содержит уникальный набор белков. В состав наружной мембраны входит белок порин, который образует широкие гидрофильные каналы в липидном бислое. (Максимович, 2015). В результате эта мембрана напоминает сито, проницаемое для всех молекул массой менее 10000 дальтон, в том числе низкомолекулярных. Эти молекулы могут проникать в межмембранное пространство, но большая их часть не способна проходить через непроницаемую внутреннюю мембрану. Основная функциональная часть митохондрии– матрикс и окружающая его внутренняя мембрана. Внутренняя мембрана содержит большое количество «двойного» фосфолипида кардиолипина (30%), что обеспечивает непроницаемость мембраны для ионов и отличается необычно высоким содержанием белка (около 70% от веса). Многие из белков являются компонентами электронтранспортной цепи, поддерживающей протонный градиент на мембране. Другой большой белковый комплекс–фермент АТФ-синтаза, катализирующий синтез АТФ, через который протоны возвращаются в матрикс по электрохимическому градиенту (Erazo-Oliveras,2014).
Роль митохондрий в энергетике клетки
Наиболее характерной особенностью митохондрий является содержание в них большого числа ферментов, участвующих в аэробном «дыхании». Большая часть энергии, которая освобождается при переносе электронов, аккумулируется в макроэргических фосфатных связях АТФ. (Максимович, 2015)
Окисление ацетильной группы в цикле Кребса ведет к образованию молекул восстановленного NADH и восстановленного FADH2. Вначале почти вся энергия, получаемая на ранних этапах окисления питательных веществ, аккумулируется в форме высокоэнергетических электронов NADH и FADH2. NADH, компонент NADH-дегидрогеназного комплекса, образовавшийся в цитозоле при гликолизе, передает свои электроны в дыхательную цепь. Так как NADH не способен проходить через внутреннюю мембрану, перенос электронов от него осуществляется непрямым путем при помощи одной из челночных систем, транспортирующих в митохондрию карнитин, который после окисления возвращается в цитозоль с последующим его восстановлением с помощью NADH. Другой субстрат, FADH2 передает свои электроны в дыхательную цепь непосредственно. Электроны этих субстратов восстанавливают молекулярный кислород (акцептор электронов) в дыхательной цепи с образованием метаболической воды. Так как большое количество высвобождаемой энергии используется ферментами внутренней мембраны для образования АТФ из AДФ, эти реакции называют окислительным фосфорилированием. На внутренней мембране создается электрохимический протонный градиент. Митохондриальная дыхательная цепь внутренней мембраны способна перемещать протоны Н+. При прохождении электронов по дыхательной цепи происходит их «откачивание» из матрикса. АТФ-синтаза может использовать энергию гидролиза АТФ для переноса Н+ через мембрану, а при достаточно большом протонном градиенте протоны начинают «течь» через фермент в обратном направлении, что сопровождается синтезом АТФ. Все белки-переносчики электронов группируются в 4 больших комплекса дыхательных ферментов, каждый из которых содержит трансмембранные белки, прочно закрепляющие комплекс во внутренней мембране митохондрии. Комплекс I (NADH-убихиноноксидоредуктаза; NADH-дегидрогеназа), комплекс II (сукцинатдегидрогеназа; сукцинат-убихинон оксидоредуктаза), комплекс III (комплекс цитохромов b, c1; убихинон-цитохром c оксидоредуктаза), комплекс IV (цитохром c оксидаза; цитохромоксидаза; цитохром с-O2 оксидоредуктаза). Каждый последующий комплекс обладает большим сродством к электронам, чем предыдущий. (Logan, 2006) Электроны последовательно переходят от одного комплекса на другой, пока не восстановят кислород, являющийся их акцептором.(Максимович, 2015)
Роль митохондрий в кальциевом гомеостазе
Центральным механизмом в реализации иммунного ответа является кальциевая сигнализация. Иммунореактивность лимфоцитов обеспечивается интеграцией митохондрий и механизмов кальциевой сигнализации. Митохондрии играют важную роль в гомеостазе Ca 2+ лимфоцитов, как и в других клетках. Они имеют огромный потенциал для его быстрого накопления, поэтому участвуют в модуляции пространственно-временного профиля кальциевых сигналов (Bonifaz 2015, Chandel 2014).
В последние годы все большее внимание исследователей привлекает изучение работы митохондрий как кальциевых депо клетки в процессе реализации специфических функций иммунокомпетентных клеток, так как белки компоненты этой сложной системы регуляции кальциевого гомеостаза могут рассматриваться в качестве молекул-мишеней для направленной регуляции функциональной активности лимфоцитов в норме и при патологических процессах (воспаление, аутоиммунная патология, аллергические реакции, иммунодефициты).
Стабильный уровень Ca 2+ в митохондриях сохраняется в результате равномерного накопления ионов и их высвобождении при значительном повышении уровня Ca 2+ в матриксе, за счет слаженной работы транспортной системы внешней и внутренней мембран митохондрий. Данная система включает основной канал тока Ca 2+ через наружную мембрану – потенциал-зависимый анионный канал; также систему унипорта внутренней мембраны и его молекулярные компоненты, регулирующие активность; два пути высвобождения Ca 2+ в цитозоль – H+/Ca 2+ насос и проницаемая пора мембраны митохондрий. Ток Ca2+ через потенциал – зависимый канал и систему унипорта осуществляется за счет электрохимического протонного градиента (Kaufman 2014).
Были определены белки, участвующие в контроле Ca 2+ тока сквозь внутреннюю мембрану митохондрий (Becker 2009). В частности, в 2010 г. были исследованы Na + /Ca 2+ насосы; белки – регуляторы поглощения Ca 2+ митохондриями, они получили название mitochondrial calcium uptake 1 белки –MICU1; затем были обнаружены и частично охарактеризованы потенциальные регуляторы тока Ca 2+ в митохондрии: MICUb, MICU2, MICU3, EMRE. На основании проведенных исследований сложилась более четкая картина осуществления поглощения ионов кальция митохондриями и сохранении гомеостаза Ca 2+ как внутри органеллы, так и клеточной системе, в целом (Becker 2009).
Шапероны в мембранах ЭПР и митохондрий обеспечивают физическое и функциональное взаимодействие между ЭПР и митохондриями. В формировании АММ главную роль играет глюкозо-регулирующий белок – шаперон GRP75, который содержится в большом количестве в митохондриях. Этот шаперон контролирует передачу кальциевого сигнала от ЭПР к митохондриям и индуцирует взаимодействие между фосфоинозитол3-фосфат-чувствительными рецепторами и VDAC1. В этом случае шаперон образует между мембранами ЭПР и митохондрий туннель для Ca2+, позволяя более эффективно проникать ионам из ЭПР во внешнюю мембрану митохондрий.
Роль митохондрий в апоптозе
Установлено, что основным компонентом, осуществляющим восприятие стимулов ПГК и активизирующим механизмы реализации той или иной формы ПГК, являются митохондрии. Предполагается, что на уровне митохондрий осуществляется интеграция сигналов активизирующих и подавляющих процесс ПГК, следствием чего является дальнейшая реализация программированной клеточной гибели или ее подавление.
На сегодняшний день показано существование трех основных форм программированной гибели клетки: апоптоз (I тип ПГК), аутофагия (II тип ПГК), некрозоподобная ПГК (III тип ПГК). Каждый из этих типов гибели клетки характеризуется собственными биохимическими, молекулярными и морфологическими особенностями (Бра 2005).
При апоптозе наблюдается уменьшение клетки в объеме, конденсация хроматина и фрагментация ДНК на олигонуклеосомные фрагменты. Митохондрии и рибосомы во время реализации апоптоза сохраняют в основном свою структуру и частично – функции. Заключительный этап апоптоза характеризуется разрушением цитоскелета, что приводит к сморщиванию клетки и ее фрагментации на апоптотические тельца, поглощаемые макрофагами или другими соседними клетками.
Ключевыми участниками терминальной фазы апоптотической программы являются цистеиновые протеазы – каспазы, осуществляющие деградацию белковых структур клетки и активирующие нуклеазы. (Бра 2005). Для аутофагии характерно набухание митохондрий и цистерн эндоплазматического ретикулума, увеличение аппарата Гольджи, секвестрация клеточных органелл аутофагическими вакуолями, конденсация хроматина и коллапс ядра.
Терминальным этапом аутофагии является разрушение клеточных органелл лизосомальными ферментами, следствием чего является деградация клетки. Образующийся после реализации аутофагии клеточный дебрис поглощается соседними клетками (Levine 2005). Заключительным событием в этом процессе является разрыв плазматической мембраны, способствующий излиянию содержимого клетки в межклеточное пространство, что способствует индукции воспалительной реакции.
Соотношение различных типов ПГК может варьироваться в зависимости от типа и силы воздействия стимула, активизирующего ПГК.
Важной особенностью митохондрий является способность к значительной амплификации исходящих от них стимулов, активирующих ПГК. Показано, что открытие митохондриальных пор является общим моментом в реализации механизмов всех обсуждаемых выше форм ПГК (Владимиров 2002). Образование пор в митохондриях приводит к выходу из митохондрий цитохрома С, способствующего образованию апоптосомы и активирующего каспазы. Этот процесс является основным механизмом апоптотической гибели клетки. Через открытые поры в митохондриях в цитоплазму высвобождаются также факторы, перемещающиеся в ядро и активирующие реализацию ПГК по независимым от каспаз механизмам: эндонуклеаза G и AIF, связывающий ДНК и активирующий нуклеазы и протеазы в ядре. Показано, что данные факторы принимают участие в развитии как апоптоза, так и некроза. Помимо активаторов ПГК, митохондрии также высвобождают ингибиторы белков, блокирующих ПГК (Smac/DIABLO, Omi/ HtrA2) и предшественников каспаз (прокаспаза 2, 3, 9) (Бра 2005).
К небелковым медиаторам клеточной гибели относятся ионы Ca2+, активирующие при их выходе в цитоплазму кальпаины и Ca 2+ зависимые липазы, что приводит к реализации некротической формы ПГК. Дополнительным фактором индукции ПГК является увеличение продукции компонентами дыхательной цепи митохондрий активных форм кислорода, активирующих механизмы апоптоза, аутофагии и некроза. На сегодняшний день известны митохондриальные апоптотические поры (mitochondrial apoptotic pores – MAP) и поры повышенной проницаемости или мегаканалы (permeability transition pores – РТP). Механизмом образования апоптотических пор в митохондриях является олигомеризация на митохондриальной мембране белков Bax и Bak. (Aradjomande 2005).
Существует мнение, что «выбор» клеткой активизации механизмов той или иной формы программированной гибели определяется количеством открытых пор в митохондриях. В том случае, если PTP формируются в нескольких митохондриях, в клетке активируется процесс аутофагии. Когда PTP открываются у большего числа митохондрий, в клетке инициируется апоптоз, что, вероятно, является следствием увеличения в цитоплазме количества цитохрома С и AIF. Наконец, когда в клетке практически во всех митохондриях открываются РТP, происходит разобщение окисления и фосфорилирования и интенсивный гидролиз АТФ митохондриальной АТФ-азой, активизируются механизмы некрозоподобной клеточной гибели (Guimaraes 2004). Минимальное количество открытых пор принципиально не влияет на процесс клеточной гибели, при большем количестве.
Считается, что определенное значение в реализации апоптоза и некрозоподобной ПГК имеет уровень продукции АТФ. Известно, что при низком уровне АТФ в клетке протекает процесс программированной гибели клетки по механизму некроза, достаточное энергообеспечение клетки способствует прохождению ПГК по механизму апоптоза (Buja 2005).
Установлено, что митохондрии обладают широким спектром белковых (цитохром С, эндонуклеаза G, AIF,) и небелковых факторов (ионы Ca2+, активные формы кислорода), активизирующих процесс клеточной гибели после высвобождения их в цитоплазму. В настоящее время существует аргументированная гипотеза, предполагающая, что накопление нарушений в митохондриальном геноме и прогрессирование митохондриальной дисфункции является одним из механизмов старения организма и развития различных патологических процессов.
На сегодняшний день известны митохондриальные апоптотические поры (MAP) и поры повышенной проницаемости или мегаканалы (permeability transition pores – РТP). Механизмом образования апоптотических пор в митохондриях является олигомеризация на митохондриальной мембране белков Bax и Bak. PTP формируются за счет объединения в единый комплекс АТФ –АДФ- антипортера, локализованного во внутренней митохондриальной мембране, циклофилина D, находящегося в матриксе митохондрий, и порина (voltage dependent anion channel, VDAC) – ионного канала внешней митохондриальной мембраны (Aradjomande, 2005).
Таким образом, Митохондриальный путь апоптоза предусматривает не только активацию каспаз, но и доставку в ядро клетки активных ферментов — эндонуклеазы G и апоптозиндуцирующего фактора, способных вызвать деградацию генетического материала без активации каспаз (Kaufman 2014).
Болезни и изменения клеточного метаболизма
Автор
Редакторы
Нейродегенеративные и онкологические болезни — самые распространенные возрастные патологии после болезней сердца и сосудов. Как показывают исследования, эти патологии тесным образом связаны с энергетическим обменом и митохондриальной дисфункцией. Детальное и масштабное изучение изменений клеточного метаболизма при развитии этих патологий способствует разработке более совершенных диагностических инструментов, позволяющих обнаруживать заболевание на самой ранней его стадии.

Биоэнергетика
Спецпроект о клеточном энергетическом метаболизме, работе митохондрий и АТФ, а также о заболеваниях, связанных с нарушениями функций клеточных «батареек».
Спонсор спецпроекта — «БиоХимМак» — поставщик научного и медицинского оборудования в лаборатории России и стран СНГ.
Наверное, у каждого, кто начинает знакомиться с удивительной организацией наших клеток, возникает чувство восхищения невероятной сложностью внутриклеточного мира. Каждую секунду в миллиардах наших клеток протекают сложные и строго скоординированные процессы. И одним из таких очень важных процессов является производство в митохондриях главной энергетической молекулы — аденозинтрифосфата, или АТФ. Сегодня уже хорошо известно, что работа митохондрий очень тесно связана со здоровьем и продолжительностью жизни [1]. Митохондрии производят энергию для поддержания жизни, но при этом они же служат основными источниками активных форм кислорода, избыток которых для клеток губителен.
Энергетический обмен
Любой живой организм находится в постоянной связи с окружающей средой, непрерывно обмениваясь с ней веществом. В этом процессе можно выделить три этапа:
Внутриклеточный метаболизм, в свою очередь, включает в себя два типа реакций: катаболизм и анаболизм.
Катаболизм — это процесс расщепления и окисления органических молекул, приводящий к образованию тепла и энергетических молекул, АТФ. Именно за счет постоянного производства—расщепления последних съеденные нами калории направляются «по адресу»: гидролиз двух высокоэнергетических (макроэргических) связей в молекулах АТФ обеспечивает энергией всевозможные синтетические и транспортные процессы в клетках.
На первом этапе катаболизма под воздействием пищеварительных ферментов сложные органические соединения (белки, полисахариды, жиры) распадаются на более простые — аминокислоты, моносахариды, жирные кислоты и глицерин, — которые клетка использует для реакций анаболизма (пластического обмена) и получения энергии. Аминокислоты идут на синтез белков. Жирные кислоты выполняют энергетическую функцию, входят в состав клеточных мембран и служат субстратом для синтеза эйкозаноидов
На втором этапе происходит гликолиз — расщепление молекул глюкозы (рис. 1) до пировиноградной кислоты (ПВК). Дальнейший ход реакций зависит от присутствия или отсутствия кислорода в клетке. Если кислорода нет (анаэробный процесс), то ПВК у микроорганизмов и растений будет превращаться в этанол, а в организме животных — в молочную кислоту [2]. Каждый, кто подвергал себя тяжелым физическим нагрузкам, мог почувствовать конечный результат анаэробного метаболизма в виде боли и скованности в мышцах из-за скопившейся в них молочной кислоты.
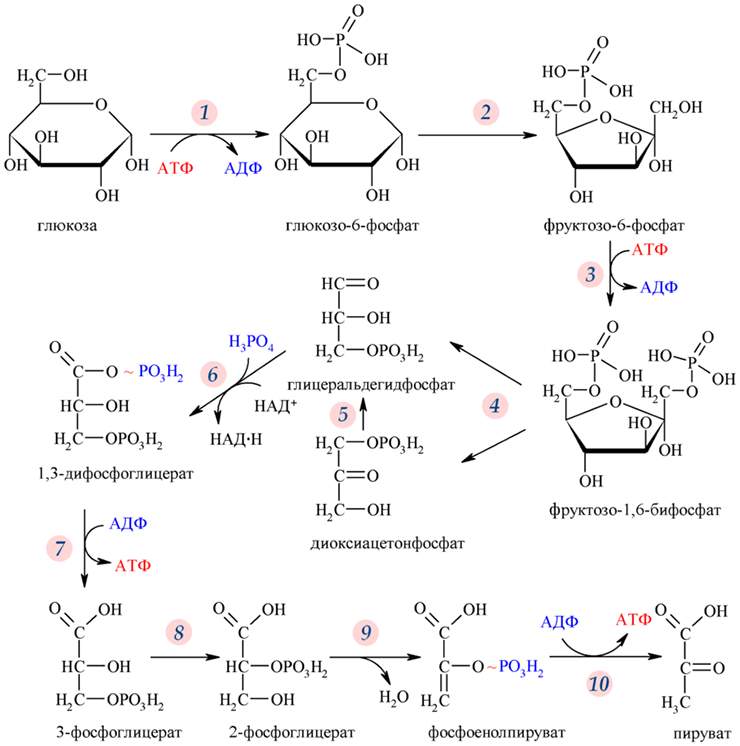
Рисунок 1. Реакции гликолиза. На 10 этапах гликолиза (пяти подготовительных и пяти этапах синтеза АТФ) из шестиуглеродной молекулы глюкозы образуются две трехуглеродные молекулы пировиноградной кислоты. Полученная от расщепления глюкозы энергия запасается в «энергетической валюте» клетки — двух молекулах АТФ и двух молекулах НАДФ.
Если же кислород в клетке есть, ПВК будет расщепляться на углекислый газ и воду и тоже высвобождать заключенную в углеводной молекуле энергию. Этот процесс называется аэробным клеточным дыханием и проходит в специальных органеллах — митохондриях. Окисление в митохондриях дает гораздо больше энергии, чем гликолиз.
Митохондрии и производство АТФ
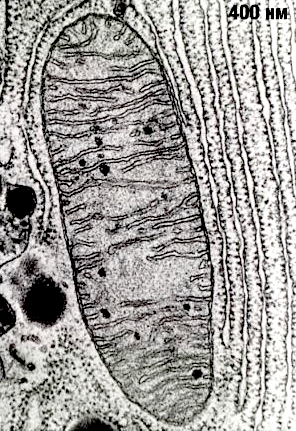
Рисунок 2. Митохондрия под электронным микроскопом.
Митохондрии — настоящее биологическое чудо, сотворенное эволюцией. Несмотря на очень маленький размер (в одной клетке может быть более 1000 митохондрий), эти органеллы поражают чрезвычайно сложной организацией (рис. 2). Они представляют собой вытянутые «пузырьки», окруженные двумя мембранами. Считается, что митохондрии сформировались в результате поглощения археями-фагоцитами пурпурных фотосинтезирующих бактерий, которые, приспосабливаясь к избытку кислорода, освоили аэробное дыхание [3], [4]. Мембраны митохондрий состоят из липидов и гидрофобных, нерастворимых в воде белков. (Здесь мы так подробно описываем строение митохондрий не случайно, а для того чтобы потом было понятно, как их нормальная работа и дисфункция влияют на здоровье.)
Строение мембран очень важно для процесса дыхания. Внешняя мембрана митохондрий — гладкая, а внутренняя — многократно складчатая. Эти складки (или кристы) позволяют увеличить рабочую площадь мембраны, что необходимо для размещения там всего комплекса белков, осуществляющих дыхание. Вначале окисляются углеродные атомы углеводов, жирных кислот и аминокислот до СО2 (гликолиз, цикл Кребса и β-окисление жирных кислот), а полученные таким образом электроны используются для образования НАДФ. Далее НАДФ окисляется молекулярным кислородом с образованием воды. НАДФ-оксидазная реакция сопровождается выделением очень большого количества свободной энергии (около 1,1 эВ при переносе одного электрона с НАДФ на кислород), которая может запасаться дыхательной цепью в виде трансмембранной разности электрохимических потенциалов ионов H+ (протонов).
Работа же дыхательных белков-ферментов похожа на работу насосов: передавая электроны друг другу, они перекачивают протоны в межмембранное пространство (см. видео 1). В результате внутренняя мембрана митохондрии заряжается подобно конденсатору. Создаются потенциалы: электрический (положительные заряды — снаружи митохондриальной мембраны, отрицательные — внутри органеллы) и химический (возникает разница концентраций протонов: внутри митохондрии их меньше, снаружи — больше). Известно, что электрический потенциал на мембране митохондрий, которая служит хорошим диэлектриком, достигает 200 мВ при толщине мембраны всего 10 нм [5]. Для сравнения: потенциал действия на мембранах нервных клеток при передаче сигнала достигает всего 30 мВ.
Видео 1. Как работает митохондрия
Накопившись в межмембранном пространстве, протоны, подобно электрическому току, устремляются назад, в митохондрию — туда, где их концентрация ниже. Однако они могут проходить только по специальным каналам АТФ-синтазы, встроенной во внутреннюю мембрану: протонный канал (ротор) этого фермента закреплен в мембране, а каталитический комплекс торчит внутрь митохондрии, в матрикс (рис. 3). Поток протонов раскручивает ротор, как река водяную мельницу. В результате ротор вращается с невероятной скоростью — 300 оборотов в секунду (см. видео 2)! И именно это вращение приводит к образованию высокоэнергетической молекулы — АТФ [6]. Подсчитано, что в сутки в организме взрослого человека синтезируется и расходуется около 40 кг АТФ, при этом жизнь каждой молекулы очень коротка.
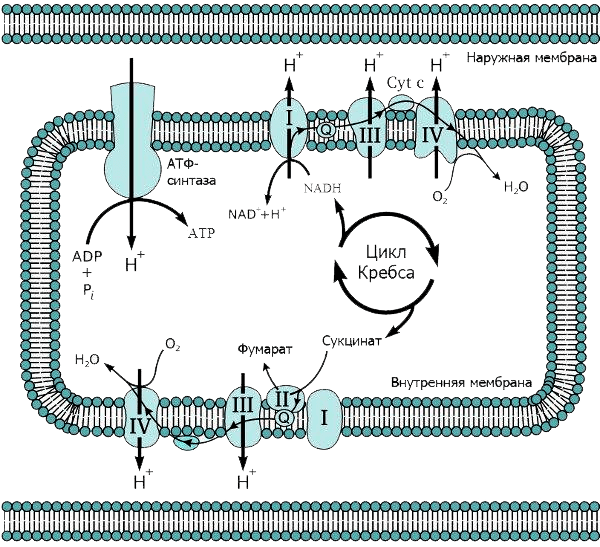
Рисунок 3. Схема дыхательной цепи митохондрий.
Видео 2. Работа АТФ-синтазы в мембране митохондрии
Всё вышесказанное имеет самое непосредственное отношение к старению. Дело в том, что в процессе дыхания ферменты работают не совсем «чисто», и в результате образуются побочные продукты — активные формы кислорода (АФК). Пока человек молод и здоров, образующиеся в митохондриях АФК не представляют для него ощутимой угрозы, так как легко нейтрализуются организмом. Но когда человек стареет, ведет нездоровый образ жизни или имеет генетическую предрасположенность к определенным болезням, его защитные системы дают сбой, рушась одна за другой.
Жирные кислоты и дисфункция митохондрий
То, что старение и возрастные патологии сопровождаются дисфункцией митохондрий, которые начинают производить меньше АТФ и хуже обновляться, уже ни у кого не вызывает сомнения. Выяснилось также, что дисфункция митохондрий и старение тесным образом связаны с повышением уровня свободных жирных кислот в крови [7], чему сильно способствуют малоподвижность и нерациональное питание. Жирные кислоты, попадая в клетку, способны напрямую снижать синтез АТФ, разобщая окисление и фосфорилирование. Этот связанный с терморегуляцией организма феномен был открыт еще шесть десятилетий назад академиком Скулачевым и его коллегами [8]. Снижение синтеза АТФ, в свою очередь, запускает сразу несколько негативных цепных реакций, связанных с возрастными болезнями и старением в целом.
И вот что происходит. Повышение уровня свободных жирных кислот в организме приводит к резистентности к инсулину: инсулинзависимые клетки перестанут реагировать на этот гормон. В результате нарушается усвоение глюкозы и жирных кислот, ухудшается окисление последних. Дело в том, что характерный для состояния инсулинорезистентности высокий уровень инсулина активирует целый каскад реакций, который блокирует работу фермента карнитинпальмитоилтрансферазы I (СРT1), участвующего в переносе жирных кислот внутрь митохондрий [9]. Из-за этого ухудшается синтез АТФ, а жирные кислоты накапливаются в цитоплазме клеток, вызывая эффект липотоксичности. Кроме резистентности к инсулину, избыток жирных кислот в организме вызывает резистентность к еще одному «пищевому» гормону — лептину. А из-за этого страдает функция одного из главных участников биогенеза (обновления) митохондрий — коактиватора рецептора гамма, активируемого пролифераторами пероксисом (PGC-1α). В итоге митохондрии производят меньше АТФ, стареют, погибают и провоцируют гибель клеток путем апоптоза [10].
Ну и наконец, избыток жирных кислот вызывает стресс эндоплазматического ретикулума (ЭПР) — внутриклеточного органоида, участвующего в синтезе белков и множестве других процессов. При стрессе ЭПР в цитоплазму высвобождаются ионы кальция, способные вызывать дисфункцию и гибель митохондрий [11]. Ионы кальция могут накапливаться в клетке и по другой причине — из-за ухудшения работы ионных насосов, откачивающих кальций из клетки. А причиной этому служит нарушение работы митохондрий, сопровождающееся снижением синтеза АТФ, без которого отказываются работать ионные насосы. В итоге формируется порочный круг: снижение выработки АТФ приводит к дисфункции митохондрий, что еще больше снижает выработку АТФ, и т.д.
Жирные кислоты, церамиды и повреждения нейронов
Как выяснилось, избыток жирных кислот и дисфункция митохондрий напрямую связаны с возникновением возрастных нейродегенеративных патологий. Надо сказать, что клетки нервной системы — самые уязвимые для возрастного окислительного стресса и снижения синтеза АТФ. Такая исключительная чувствительность нейронов к дефициту энергии и повышению генерации АФК объясняется несколькими причинами.
Во-первых, нервная ткань в силу своей физиологии нуждается в наибольшем потреблении кислорода. Вследствие этого в митохондриях нейронов происходит интенсивный окислительный метаболизм, который и становится основной причиной повышенной генерации АФК.
Во-вторых, из-за того, что мембраны нейронов содержат много ненасыщенных жирных кислот, они легко подвергаются перекисному окислению липидов. Так как активность антиоксидантных систем в ткани головного мозга ниже, чем в других органах, а с возрастом сокращается и количество некоторых ферментов-антиоксидантов, становится понятным, почему клетки нервной системы наиболее чувствительны к окислительным повреждениям [12].
В настоящее время известно несколько факторов, повреждающих нейроны. Среди них — белки, образующие внутри- и внеклеточные агрегаты (β-амилоидный белок и другие), а также церамиды и липофусцин. На их количество влияет прежде всего избыток жирных кислот в организме. Отягчающим обстоятельством в этом случае выступает чрезмерное содержание насыщенных кислот (пальмитиновой и стеариновой) в пищевом рационе. Всё это вместе служит мощным стимулом развития разнообразных нейродегенеративных заболеваний, таких как болезнь Альцгеймера [13], [14].
Но каким же образом пальмитиновая кислота может способствовать нейродегенерации? Установлено, что из-за избытка этой кислоты накапливаются церамиды, которые участвуют в регуляции терминальной дифференцировки, пролиферации и апоптоза нейронов. Посредством нескольких химических реакций они воздействуют на регуляторы клеточного цикла, повышая концентрацию ингибиторов киназ p21/SDI1 и p27/KIP1. Таким образом церамиды останавливают клеточный цикл, что, в свою очередь, активирует главного «стража генома» — белок р53 — и «насылает» на клетку апоптоз [15]. Кроме этого, при деградации церамида образуется вещество сфингозин, обладающее цитотоксическим действием и способное вызывать как апоптоз, так и некроз клеток. Но и это еще не всё. Обнаружено, что накопление насыщенных жирных кислот (пальмитиновой и стеариновой) стимулирует специальные клетки головного мозга (астроглию) на эндогенный (внутренний) синтез церамидов. Эти произведенные церамиды запускают цепную реакцию следующего вида: церамиды → повышение секреции провоспалительных цитокинов и оксида азота → увеличение производства АФК и окислительный стресс → активация стресс-регулируемых киназ (CDK5 и GSK-3) в нейронах → образование β-амилоидного белка и гиперфосфорилирование τ-белка [16].
Нейродегенеративные патологии и дисфункция митохондрий
Сегодня важнейшими и самыми распространенными нейродегенеративными патологиями считают болезни Альцгеймера, Паркинсона, Хантингтона, а также боковой амиотрофический склероз. Их возникновение связывают со структурными изменениями различных белков, приводящими к образованию внутриклеточных агрегатов. К таким белкам относятся:
Болезнь Альцгеймера (БА) — тяжелое нейродегенеративное заболевание, для которого характерны синаптическая дисфункция и гибель нейронов, что сопровождается снижением когнитивных способностей: ухудшением памяти и мышления, постепенной потерей социальных и моторных навыков [17]. В зоне риска развития болезни находятся в основном пожилые люди. Лишь 1–2% людей в возрасте до 65 лет страдают БА. Согласно одной из гипотез развития БА — амилоидной, — болезнь возникает из-за накопления в головном мозге агрегатов β-амилоида. Этот пептид состоит из 39–43 аминокислотных остатков и является фрагментом крупного трансмембранного белка под названием предшественник бета-амилоида (amyloid precursor protein, APP). Находясь в избытке, молекулы β-амилоида начинают «склеиваться» и образовывать нерастворимые бляшки (рис. 4). Именно в таком состоянии белок нарушает работу нервных клеток и вызывает симптомы БА. У страдающих БА в пораженных участках мозга находят большое количество амилоидных бляшек и нейрофибриллярных клубков [18].
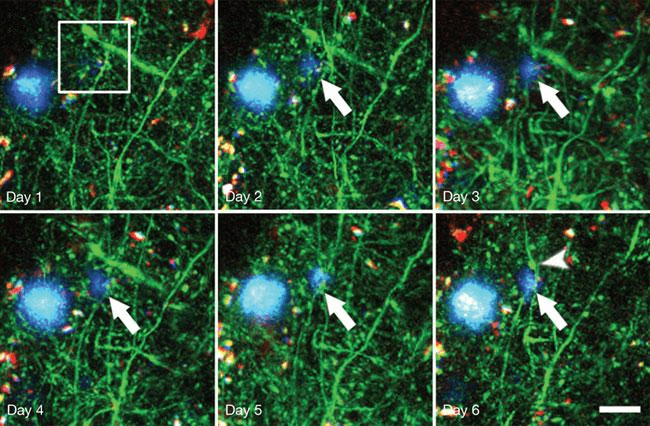
Рисунок 4. Образование амилоидной бляшки у генно-инженерных мышей (показано длинной стрелкой). На 6-й день уже видна дистрофия нейрона (короткая стрелка). Синим цветом обозначены отложения амилоида, зеленым — нейроны. Длина масштабной линейки — 20 мкм; снимки сделаны с помощью мультифотонного микроскопа.
Однако амилоидная гипотеза — не единственная, объясняющая возникновение БА. В 1993 году Аллен Роузес, профессор Университета Дьюка, предложил еще одну гипотезу возникновения БА — генетическую, связанную с геном APOE, кодирующим аполипопротеин Е (ApoE). Выяснилось, что наследование одного из вариантов гена APOE — APOE4 — в несколько раз повышает шансы заболеть БА. Всё больше исследователей склоняются к мысли, что β-амилоид излишне «демонизирован» и не является первопричиной развития БА. Неудавшаяся терапия, направленная на очистку клеток от β-амилоида, подтверждает, что с этой болезнью не всё до конца ясно [19].
Болезнь Паркинсона (БП) — еще одно тяжелое и довольно распространенное возрастное нейродегенеративное заболевание. У больных БП в нейронах черной субстанции накапливается α-синуклеин, который образует особые гранулы — тельца Леви. Надо сказать, что существует так называемая деменция с тельцами Леви, для которой характерно скопление многочисленных телец Леви в кортикальных и субкортикальных нейронах и развитие прогрессирующего когнитивного расстройства уже в первый год заболевания. Но пока не совсем ясно, считать ли эту деменцию формой БП или же правильнее ее рассматривать как отдельное заболевание. В случае БП скопления телец Леви приводят к дисфункции нейронов и их гибели, при этом характерно поражение областей мозга из состава так называемого нигростриарного дофаминового пути. Этот путь регулирует двигательную активность, снижая напряжение в мышцах. Вот почему, когда гибнут дофаминовые нейроны, у больных возникают соответствующие симптомы: нарастающее повышение мышечного тонуса и дрожание рук. Кроме нарушения моторных функций для БП характерны и другие симптомы, связанные с нарушением сна, депрессией, тревогой, ухудшением зрения и замедлением мышления [20].
Болезнь Хантингтона (БХ) — тоже не слишком редкое нейродегенеративное заболевание [21]. Как и в случае болезни Альцгеймера, для патогенеза БХ характерно образование токсичных белковых агрегатов с участием мутантных форм белков, которые синтезируются в нервной ткани. Но если к основному «виновнику» БА, β-амилоиду, у ученых есть вопросы, в случае с БХ сомнений гораздо меньше. Установлено, что именно генетические особенности — полиморфизмы определенных участков ДНК — приводят к появлению патологических форм белка хантингтина. Такой хантингтин способен к ассоциации с другими белками нервной ткани, в результате чего образуются нерастворимые токсичные агрегаты, повреждающие кору и полосатое тело головного мозга. Для БХ типичны всплески непроизвольной двигательной активности, эмоциональные расстройства и потеря памяти. В то же время нормальная физиологическая функция белка хантингтина в организме остается под вопросом. Предполагают, что он играет какую-то роль в эмбриогенезе [22].
Все три упомянутые патологии самым тесным образом связаны с дисфункцией митохондрий. Прежде всего, надо отметить, что ее развитие под действием дефектных белков, специфичных для нейропатологий, было установлено несколькими способами: in vitro (на клеточных линиях и внеклеточных системах) и in vivo (на трансгенных животных). Обнаружили и обратную связь: оказалось, что дисфункция митохондрий может стимулировать появление дефектных белков. Так, нарушение активности дыхательного комплекса I ведет к накоплению в нервных клетках гиперфосфорилированного τ-белка и α-синуклеина [23].
Со скоплением дефектных белков связали и уже упоминавшийся стресс эндоплазматического ретикулума. Один из таких белков, α-синуклеин, может снижать активность протеасом, что заканчивается стрессом ЭПР, увеличением производства АФК и инициацией апоптозных процессов. Это происходит потому, что из митохондрий высвобождается апоптозный фактор, цитохром С, который активирует «клеточных убийц» — каспазу-9 и каспазу-3 [24]. Как полагают, на начальных этапах нейродегенерации при БА накопление β-амилоида и гиперфосфорилирование τ-белка могут быть физиологическими механизмами защиты клетки от окислительного стресса, вызванного прогрессирующей митохондриальной дисфункцией. Однако при избыточном накоплении этих белков в клетке происходит сбой в работе митохондрий. Так, у пациентов с БА обнаружили, что β-амилоид накапливается в митохондриях и нарушает реакции гликолиза и цикла Кребса, активизирует продукцию АФК. Более того, β-амилоид способен напрямую подавлять синтез АТФ. Это возможно из-за структурного сходства белка с естественным ингибитором F(1)-субъединицы АТФ-синтазы митохондрий. Также β-амилоид может взаимодействовать с митохондриальной мембраной, формируя стабильные комплексы с двумя транслоказами, TOM40 и TIM23. Такие комплексы подавляют импорт в митохондрии белков, кодируемых ядерным геномом, — субъединиц IV и Vb цитохромоксидазы. На что органелла откликается увеличением производства агрессивного пероксида водорода.
Но и это еще не всё: белок — предшественник β-амилоида может формировать поры в мембранах митохондрий и других органелл, что нарушает ионный баланс в клетке и запускает ее апоптоз [25]. Также этот белок повышает активность фосфолипазы D, в результате изменяя фосфолипидный состав митохондриальных мембран, увеличивая концентрацию фосфатидилхолина, фосфатидилэтаноламина и фосфатидной кислоты и нарушая работу мембран. Известно, что β-амилоид может связывать гем, а это ведет к дефициту гема в клетке, из-за чего нарушается работа гем-содержащего IV комплекса электронтранспортной цепи митохондрий [26].
Но не только β-амилоид способен негативно влиять на митохондрии. В экспериментах с трансгенными грызунами, экспрессирующими ген хантингтина человека, обнаружили агрегацию этого белка в митохондриях с последующим развитием их дисфункции. Другой «зловредный» белок, α-синуклеин, накапливаясь во внутренней митохондриальной мембране, способен снижать активность дыхательного комплекса I. Как следствие, митохондрии увеличивают продукцию АФК [27]. Также обнаружено, что α-синуклеин, взаимодействуя с митохондриями, может стимулировать высвобождение из них цитохрома С, а значит, инициировать апоптоз.
В целом, можно сказать, что запуск апоптоза — характерный эффект белков, вызывающих нейродегенерацию. Они могут прямо или косвенно воздействовать на регуляторные белки, связанные с апоптозом: p53, Akt, Bad, Bax, Bcl-x(L) и кальцинейрин [28].
Также описано, что сверхсинтез белка — предшественника β-амилоида приводит к повреждению системы слияния—деления митохондрий. Негативно влияют на эту же систему и на утилизацию дефектных митохондрий аутофагосомами мутации гена паркина (PARK2), обнаруженные у больных БП. Дефектные формы τ-белка и хантингтина тоже мешают нормальной работе митохондрий, ухудшая тем самым энергообеспечение отростков нервных клеток и синаптическую передачу, вызывая дегенерацию синапсов [29].
Таким образом, белки, участвующие в развитии нейродегенеративных патологий, могут способствовать митохондриальной дисфункции посредством целого ряда механизмов. В свою очередь, уже возникшая дисфункция может усугублять патологические процессы, стимулируя появление дефектных белков и замыкая тем самым порочный круг развития болезни.
Эффект Варбурга
И напоследок стόит коснуться еще одного момента, связанного с патологиями и изменением клеточного метаболизма. В 1926 году немецкий биохимик Отто Варбург сравнил скорости образования молочной кислоты (лактата) в нормальных и опухолевых клетках. Оказалось, что опухолевые клетки потребляют очень много глюкозы, образуя при этом лактат. И делают это они гораздо быстрее, чем нормальные клетки: злокачественная ткань в эксперименте производила молочную кислоту в восемь раз активнее, чем это происходит в мышце, выполняющей физическую работу. Варбург установил, что раковые клетки используют гликолиз для получения энергии вне зависимости от доступности кислорода (рис. 5) [30]. В честь первооткрывателя этот феномен назвали эффектом Варбурга [2].
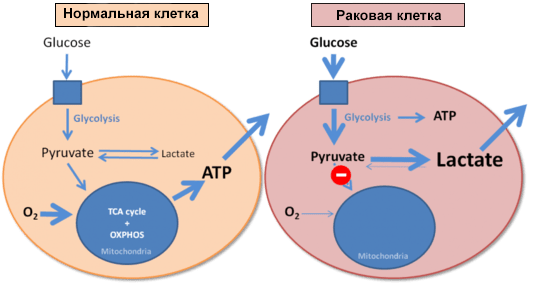
Рисунок 5. Энергообеспечение нормальной и раковой клеток. Синим квадратом обозначена поступающая в клетку глюкоза.
Обнаружив этот эффект, Варбург логично предположил, что его можно объяснить дисфункцией митохондрий в опухолевых клетках и нарушением окислительного фосфорилирования. Сегодня эта точка зрения ставится под сомнение, так как и в перерожденной ткани обнаруживают большое количество нормально работающих митохондрий. Около половины всей энергии опухолевые клетки получают из молекул АТФ, произведенных в митохондриях [31]. Эффект Варбурга проявляется в клетках уже в самом начале их трансформации в опухолевые. И это дает возможность проводить раннюю диагностику неопластических процессов: как только клетка начала расходовать глюкозу в повышенных масштабах, пора бить тревогу. Обнаружить эти процессы можно с помощью позитронно-эмиссионной томографии с использованием фторированного аналога глюкозы, 2-( 18 F)-2-дезокси-D-глюкозы.
Но зачем раковые клетки переходят на анаэробный гликолиз? Сейчас считается, что так они получают преимущество, заранее подготавливаясь к «тяжелым временам» — развитию гипоксии. А кроме этого, такой способ энергообеспечения дает клеткам возможность использовать промежуточные продукты гликолиза для анаболических реакций, усиления своей антиоксидантной защиты и отражения иммунной атаки организма [32].
Таким образом, изменения в метаболизме глюкозы и появление дефектных белков и внутриклеточных агрегатов могут говорить о начале развития патологии. Своевременное выявление подобных внутриклеточных процессов может сыграть решающую роль в предупреждении и терапии самых распространенных нейродегенеративных и онкологических заболеваний. А для того чтобы это было возможным, необходимо изучать фундаментальные аспекты патологий, связанные с работой митохондрий и энергетическим обменом. Сегодня уже разработаны системы, позволяющие заглянуть «вглубь» этих заболеваний и даже провести диагностику на самой ранней стадии их развития. Подробнее об этих системах, принципах их действия и исследованиях с их использованием расскажут следующие статьи спецпроекта.
ЗАО «БиоХимМак» — спонсор спецпроекта по биоэнергетике
Компания более 25 лет успешно занимается поставками научного и медицинского оборудования российских и зарубежных производителей: Beckman Coulter, Bio-Rad, Molecular Devices, Thermo Fisher Scientific, UVP, Seahorse Bioscience (part of Agilent), Immucor, MRC Holland и др. «БиоХимМак» обслуживает более 5000 научных и медико-диагностических лабораторий в России и странах СНГ.
Отдел молекулярной диагностики (Life Science MDx)
Молекулярная онкология, преимплантационный скрининг, цитогенетика, пренатальные и постнатальные исследования, диагностика инфекций, наследственных, мультифакторных заболеваний, детекция генномодифицированных источников и бактериального загрязнения в продуктах питания, криминалистические приложения — это лишь неполный перечень областей, которые входят в сферу интересов отдела.
Основные направления деятельности отдела:
Отдел работает как с инновационной продукцией (MLPA, PGS и NGS исследования, клеточная биоэнергетика Agilent Seahorse Bioscience), так и с зарекомендовавшими себя мировыми брендами — Beckman Coulter, Bio-Rad, Molecular Devices, UVP, Thermo Fisher Scientific.
Материал предоставлен партнёром — компанией «БиоХимМак»



